Research
Despite having sequenced the human genome over fifteen years ago, much is still unknown about how it functions. With the advent of high-throughput genomics technologies, it is now possible to measure properties of the genome across the entire genome in a single experiment, such as measuring where a given protein binds to the DNA or what genes are expressed. However, the complexity and massive scale of these data sets—billions of base pairs with thousands of measurements each—pose challenges to their analysis. My research focuses on the development of new machine learning methods that address the challenges posed by genomics data sets.
Chromatin state annotation
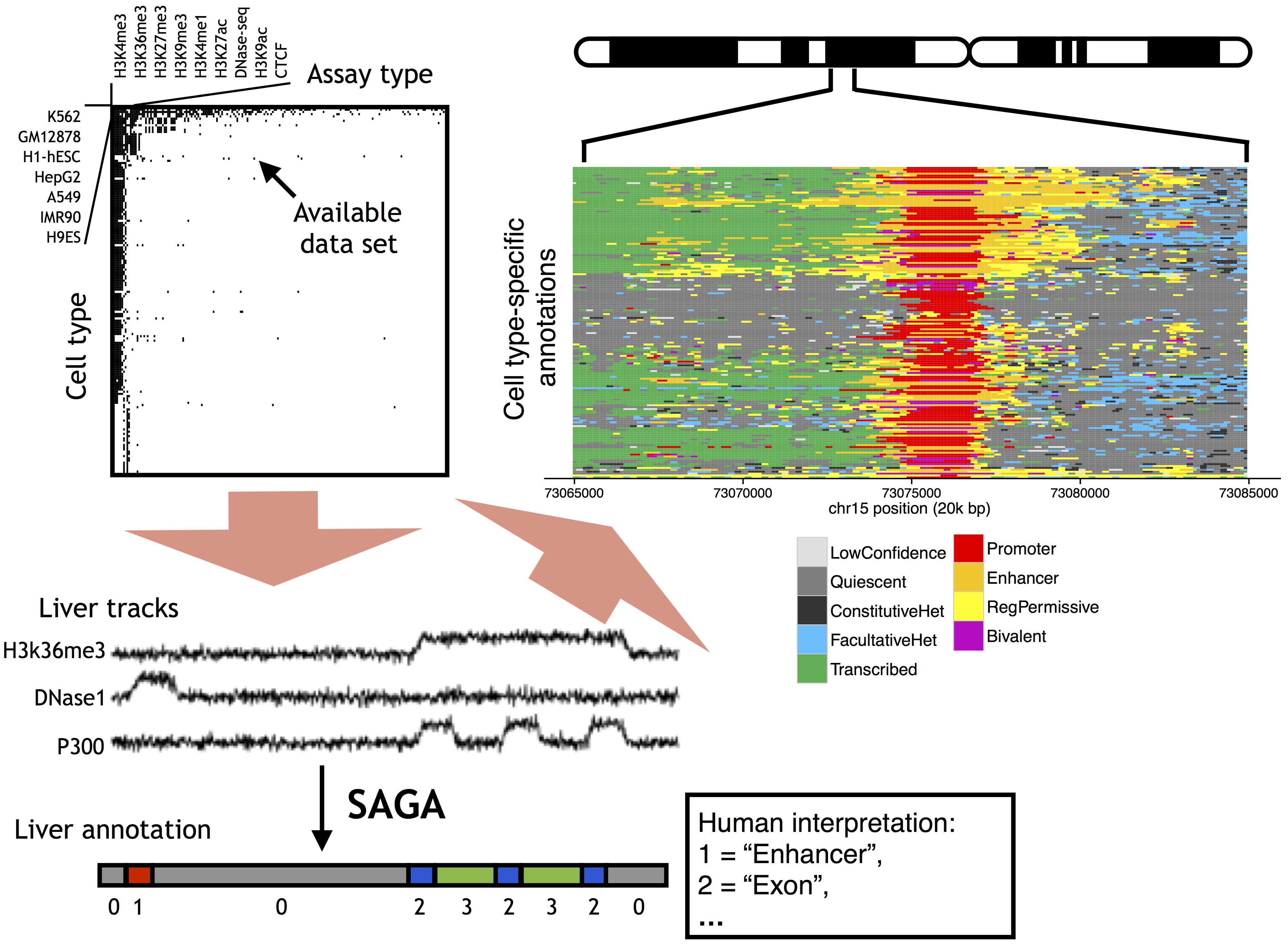
The ENCODE Project was founded with the goal of creating a catalog of functional elements in the human genome.
To that end, ENCODE and other consortia have generated thousands of genomics assays from hundreds of human tissue and cell types.
I am interested in developing integrative machine learning methods that use these genomics assays to discover and catalog functional activity in the human genome.
I help develop and maintain the Segway method for genome annotation, in collaboration with
Relevant publications:
- Michael M. Hoffman*, Jason Ernst*, Steven P. Wilder, Anshul Kundaje, Robert S. Harris, Maxwell W. Libbrecht, Belinda Giardine, Jeffrey A. Bilmes, Ewan Birney, Ross C. Hardison, Ian Dunham, Manolis Kellis, and William Stafford Noble. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Research, 41(2):827--841, 2013. Featured article.
- Maxwell W. Libbrecht*, Oscar Rodgriguez*, Jeffrey A. Bilmes, Zhiping Weng, William S. Noble. A unified encyclopedia of human functional elements through fully automated annotation of 164 human cell types.
Genome architecture and chromatin domains
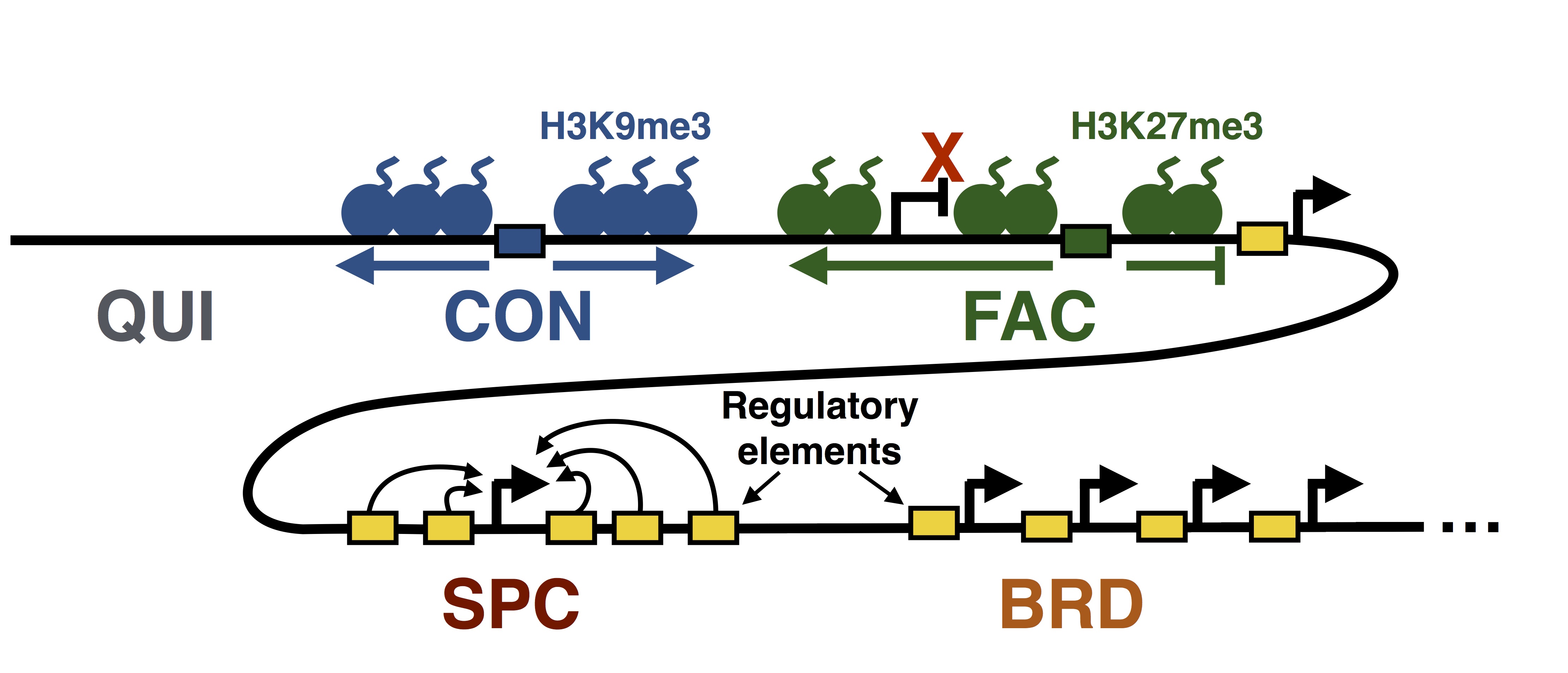
The genomic domain where a gene resides (on the scale of 100k-1M base pairs) influences its regulation: the same gene with the same local regulatory elements (that is, the same promoter) may be expressed in one domain but be silent in another. This type of regulation is crucial for gene regulation, but is currently much less well understood than local regulation. The recent availability of sequencing-based genomics assays provide a new way to understand this type of regulation. In particular, high-throughput 3C-based assays can measure the 3D conformation of DNA in the nucleus, which is known to play a key role in large-scale regulation. I am interested in developing computational methods that help us understand this aspect of genome biology.
Relevant publications:
- Maxwell W. Libbrecht, Ferhat Ay, Michael M. Hoffman, David M. Gilbert, Jeffrey A. Bilmes, and William S. Noble. Joint annotation of chromatin state and chromatin conformation reveals relationships among domain types and identifies domains of cell-type-specific expression. Genome Research, 25: 544-557, 2015. Named one of ISCB's Top 10 Regulatory and Systems Genomics papers of 2015.
Probabilistic graphical models and posterior regularization
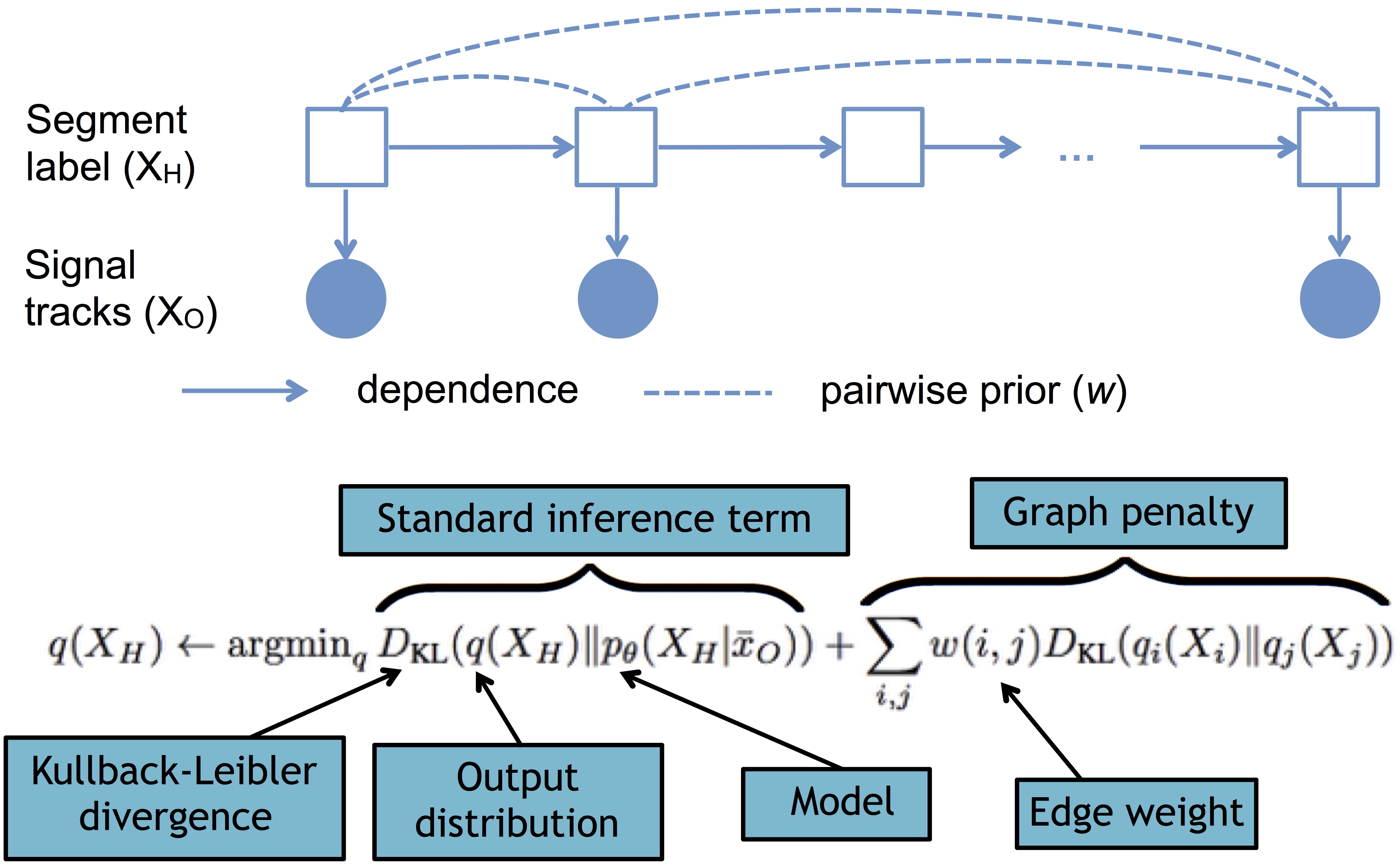
Probabilistic graphical models provide a way to encode the structure of statistical dependency between random variables. These methods can allow for efficient probabilistic inference and therefore are widely used throughout biology and other fields. I am interested in developing new methods for probabilistic graphical modeling, and in particular in the method of posterior regularization. Regularization is a concept from optimization, in which terms are added to an objective function (the target of optimization) in order to encourage the solution to have some desired property. Posterior regularization is a method for applying this concept of regulariztion to probabilistic modeling. Using posterior regularization can often be more efficient and intuitive than trying to achieve the same result by modifying the probabilistic model itself.
Relevant publications:
- Maxwell W. Libbrecht, Michael M. Hoffman, Jeffrey A. Bilmes, William S. Noble. Entropic graph-based posterior regularization. Proceedings of the International Conference on Machine Learning (ICML) 2015.
Representative set selection using submodular maximization
Convex optimization has revolutionized many fields in the past few decades, including machine learning and computational biology. Submodularity is a discrete analog to convexity: a set function—that is a mathematical function defined on subsets of a larger set—is submodular if it satisfies a particular diminishing returns property. Submodular optimization has had great success in many fields, but it is not yet widely used in biology. Recently, new methods have been developed for efficient approximate submodular maximization. I am interested in applying submodular maximization to biological problems, particularly for the task of selecting representative subsets of biological data sets.
Relevant publications:
- Kai Wei*, Maxwell W. Libbrecht*, Jeffrey A. Bilmes, William S. Noble. Choosing panels of genomics assays using submodular optimization. Genome Biology (in press).
- Maxwell W. Libbrecht, Jeffrey A. Bilmes, William S. Noble. Eliminating redundancy among protein sequences using submodular optimization. Submitted.